A devastating, once-in-a-century pandemic unleashed by COVID-19 has launched a groundbreaking vaccine development effort at a global scale: one never been seen before in the history of modern medicine. By June 2020, up to 133 candidate vaccines— 10 of which are in clinical trial —have been listed by the World Health Organization (WHO) undergoing current testing against SARS-CoV-2, the causal pathogen of COVID-19.1 To reach such extent in an exceedingly short time, only months after the first virus isolation in Wuhan,2 is unprecedented among vaccine studies. For example, no licensed vaccines are available for Zika and respiratory syncytial (RS) virus despite 70 years of research. Scientists are also well-aware, however, of the sheer difficulty in finding an effective vaccine for epidemic-causing viruses such as the human immunodeficiency virus (HIV) or dengue. Thus, realistically speaking, it might take one year until COVID-19 vaccine is ready for licensed use even galvanized by the current global effort.3 Nonetheless, most experts agree that a safe and effective vaccine is the most affordable long-term solution to induce herd immunity and suppress recurrent waves of infection.4 Fortunately, vaccine development had a good head start owing to research in viruses of the same family, SARS-CoV-1 (the cause of 2003 SARS pandemic) and MERS-CoV (the cause of 2012 pneumonia epidemic in the Middle East). These studies led to the robust consensus that the protruding crowns on the viral surface, known as spike proteins, are highly likely to induce potent antibodies.5–7
There is a sensible concern, however, surrounding whether a vaccine could be effective and applicable to all COVID-19 cases. Current vaccine development efforts are focused on China, United States, and Europe. Could any vaccine produced there be used in other parts of the world? This becomes more relevant for developing nations, especially those in Southeast Asia (SEA), where medical research is often carried out under restrained budgets and limited facilities. Vaccine research and development (R&D) is very costly to do, and any fruitful results under such constrained settings would be delayed at best. It is thus understandable that pressure is growing— voiced even by SEA government leaders 8,9—to fully focus on domestic COVID-19 vaccine research and even disregard progress made elsewhere. What is of concern here is whether the virus is so different between countries that a “foreign” vaccine would be useless. And is the virus evolving so quickly that it could resist vaccines, or even become more threatening as time passes? To answer these, we need to look into the genome of SARS-CoV-2, and understand the nature of viral mutation. Specifically, in this article I will broadly address the SARS-CoV-2 mutation rate, viral genomic distribution both globally and in SEA countries in particular, and implications for a future vaccine.
How fast does the SARS-CoV-2 mutate?
Mutation is the changing structure of genes. We can think of genes as sequences of four letters (A, C, T, and G; nucleotides) and a virus as an organism so primitive that it is just letters wrapped in an envelope (or technically a capsid). Viruses copy-and-paste these letters inside hosts’ cells to reproduce, but because the copy mechanism is not accurate, the texts naturally change over time — ergo mutation. Importantly, mutation is deemed to be one characteristic of the virus that determines whether an effective vaccine can be produced. Indeed, vaccine development has been notoriously difficult for HIV and hepatitis C virus (HCV), two RNA viruses that mutate faster than most. A high rate of mutations creates a genetically varied population of viruses (technically known as quasispecies), generating diverse subtypes with differing geographical distributions. For instance, HIV and HCV have 9 and 7 subtypes respectively, with numerous recombinations between them; HIV-01AE and HCV-1b are currently the most prevalent in SEA. Influenza viruses’ RNA and subtypes change so rapidly that we need to update the vaccine design every year. Mutations in dengue virus (DENV), another RNA virus, have rendered four serotypes that are perpetually ravaging SEA. Although not as genetically diverse as HIV, designing a vaccine with a wide enough coverage for these four DENVs has proven to be an arduous task so far.
RNA viruses are noteworthy quick mutators, due to their high tendency to make errors when copying their genomes. For viruses themselves, however, mutation is a double-edged sword — it may help them adapt to a new cellular milieu and escape host immunity; or it could be harmful, reducing their capability to replicate. Most mutations are neutral and they do not alter viral proteins at all. This balance of effect determines the fate of the virus. To place this in context, the poliovirus mutates faster than HIV-1, but it does so in a highly rigid fitness constraint. In other words, more of its offspring die after mutating and this causes the virus to be much less tolerant to mutation than HIV-1.10 Polio is essentially eradicated by vaccine nowadays. Yet, curiously enough, viral mutation can benefit vaccine production. Culturing viruses dangerous to humans long enough in other hosts, such as animal cells, will conceive progenies that mutate so much that they become non-pathogenic to humans. This innovation has created powerful live attenuated vaccines (LAV) for measles and mumps viruses, to name a few.11 Consequently, it is erroneous to say that highly mutating viruses are more dangerous, or resistant to vaccines;12 but mutation does make RNA viruses to be more unpredictable.
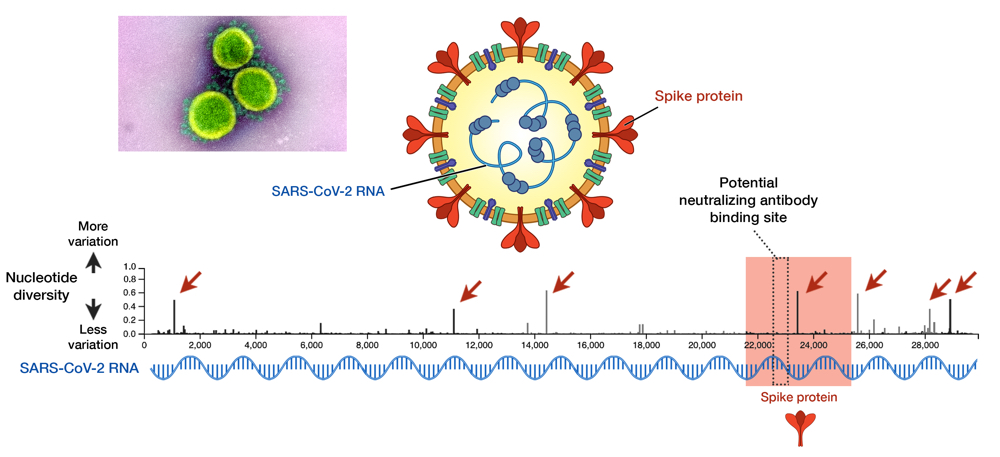
Fig 1. SARS-CoV-2 and its genome diversity. Nucleotide positions are shown on an x axis, while nucleotide diversity is shown on a y axis. Genes that encode spike proteins are highlighted in red. High diversity means high mutation at that site. Seven sites, indicated by red arrows, show a high diversity and these are called mutational hotspots. One of them (nucleotide position 23,403) is inside the gene encoding spike protein, but it is outside the antibody-binding site signified by the dotted box (position 22,559 to 23,143). Data taken from GISAID library; updated on 29 May 2020.13 Electron micrograph by NIAID.
As an RNA virus itself, it was initially feared that SARS-CoV-2 may also exhibit unpredictable mutational traits. Thus, as genomic data has been gathered and features of the viral RNA have been steadily elucidated, it is reassuring to see that mutations seem to be sufficiently contained and are likely to halt any significant changes upon the virus. The global genomic data of SARS-CoV-2, fully accessible in the GISAID repository (https://www.gisaid.org), has so far shown very little variation in its genome (Fig. 1).13 Only seven sites have shown considerable diversity within its whole genome, which stretches in length to almost 30,000 nucleotides — one of the longest among RNA viruses known to date. How diverse is that in relative to other viruses? We can juxtapose SARS-CoV-2 and the HIV-1 evolutionary tree to illustrate the difference (Fig. 2).14 To compare to HIV-1— one of the fastest mutating biological entities —might be a bit unfair, but there is confidence that all genomic isolates of SARS-CoV-2 are conserved enough and share the same antibody-binding site located within the spike protein.5,15 Furthermore, there is scarce evidence of viral adaptation to human hosts in this ongoing pandemic, and thus, SARS-CoV-2 vaccine would likely cover all circulating strains.14
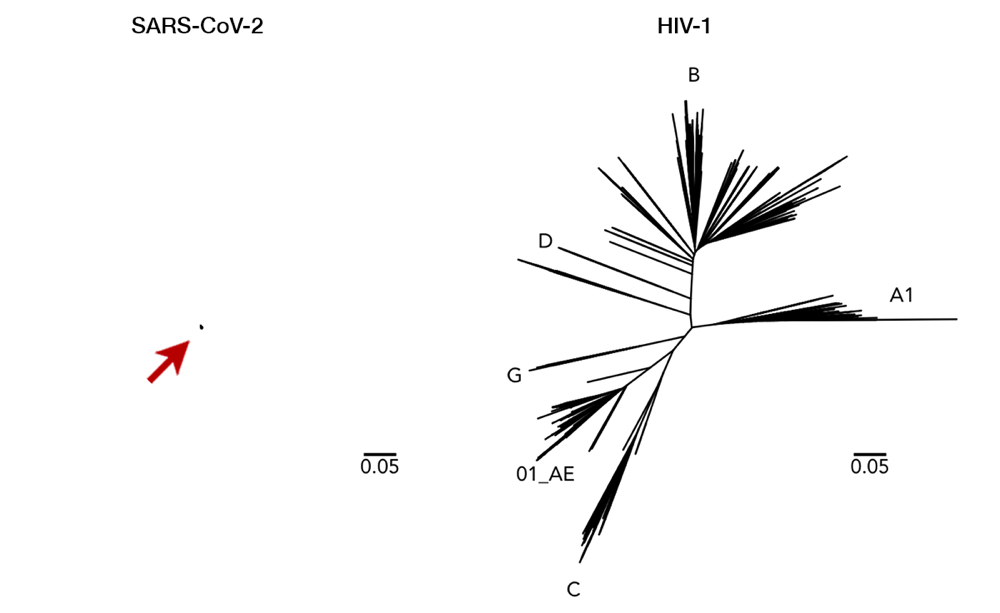
Fig 2. Here we can see a comparison of genetic diversity between SARS-CoV-2 (left, arrow for clarity) and HIV-1 (right). Longer lines mean a more distant relationship between two strains, i.e. a more mutated genome. This juxtaposition with the same scaling illustrates how miniscule the diversity of SARS-CoV-2 has been so far. While a vaccine with enormous breadth is necessary to cover all variations of HIV-1, a single vaccine is likely to cover all of SARS-CoV-2. Having said that, it is important to note that SARS-CoV-2 and HIV-1 are completely different viruses with distinct characteristics (i.e. transmission, host pathogenesis, etc). This figure is used with permission from Dr. Morgane Rolland.14
Granted, SARS-CoV-2 evolution is still in its infancy, but a molecular clock calculation so far has revealed that its mutation rate is exceedingly slow. Taking over 5,000 genomic isolates as curated by GISAID into account, the mutation rate of SARS-CoV-2 is estimated to be 8 x 10-4 substitutions/nucleotide/year.16 This translates to an 8 in 10,000 chance of mutation for every nucleotide (A-to-U, C-to-G, and so on). Approximately 25 nucleotide changes can be expected throughout its genome after one year of full circulation. For comparison, seasonal flu virus has at least 50 changes a year with only half the genome size.17 The rate of change for SARS-CoV-2 is similar to other human coronaviruses (CoVs)— both the aggressive (SARS-CoV-1 and MERS-CoV) and the mild, common cold-inducing ones (e.g. HCoV-OC43) —but much slower than other RNA viruses (Fig. 3). While RNA viruses have a much faster evolution than DNA viruses (e.g. herpes simplex virus), CoVs tend to reside in the lower register, almost similar to the measles virus — an RNA virus known for its genome stability and whose vaccine is remarkably protective. The slow evolution of CoVs mainly owe to their large genome size, and a unique RNA proofreading mechanism which ensures the fidelity of RNA synthesis during replication.18
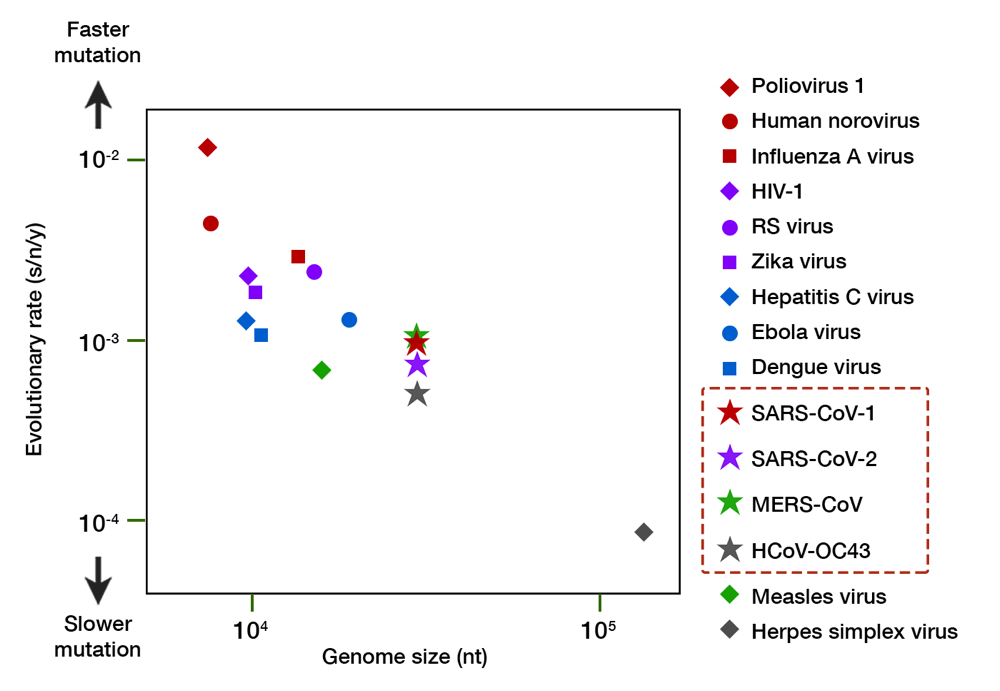
Fig 3. A comparison of the evolutionary rate of RNA viruses. The evolutionary rate (shown in substitution per nucleotide per year, s/n/y) is depicted along the y axis, and genome size (in number of nucleotides, nt) along the x axis. Organisms with a larger genome tend to have slower mutation rate. The family of coronaviruses, including SARS-CoV-2, have an enormous 30,000 nt genome and exhibit a slower mutation relative to other RNA viruses. Herpes simplex virus, a DNA virus, is shown for comparison. Data compiled from multiple sources.16,17,19–22
While signs of SARS-CoV-2 mutations so far seem unlikely to hinder vaccine binding, the threat of mutation must not be taken lightly. Recent reports have indicated the possibility of mutations altering viral pathogenicity, i.e. a capacity to inflict disease. Mutations inside the spike protein (position 23,403 in Fig. 1, also called the D614G mutation) may enhance viral binding to human ACE-2 — a human receptor used by the virus to enter cells.23 Another mutation is in polymerase (position 14,409, P323L mutation) — the viral machinery used for RNA copying. Polymerase is the target of attack of remdesivir, the sole antiviral drug currently approved for COVID-19 treatment.24 Whether these two mutations actually trigger the virus to be more deadly or drug-resistant, however, remains to be studied. One alarming caveat is that these mutant viruses began to spread in Europe in places with a high case fatality rate (CFR), before spreading to become the dominant form in many other countries.25 These mutants were not apparent at the beginning of outbreak in China. Indeed, SARS-CoV-2 mutations thus far have shed light not only on the viral evolution, but also the geographical pattern of COVID-19 transmission all over the globe. To understand that is one of the main purposes of phylogenetical study, which is discussed below.
Global transmission pattern of COVID-19: the five clades
By aligning viral genomes side-by-side, we can measure the closeness between any two strains circulating in two different places and link them to a common ancestor. Thus, we can learn which strain emerged first, construct the most likely evolutionary tree, and determine the geographical source of a specific isolate which can hint at how it was originally spread.26 For instance, an HIV-1 strain which is now widespread in SEA (subtype 01AE) was found to be genetically similar with African isolates in the 1970s — it was later known to have spread via sex workers migrating from Central Africa to Thailand at that time. Tracing mutations further back revealed the ancestor of HIV-1 (the central point in Fig. 2) to be from Kinshasa in the 1920s, transferred from chimpanzees to humans. We can trace viral mutations even further back into the past. A common ancestor of HCV was determined to be from ~1100 BC of probably SEA origin, and hepatitis B virus (HBV) is believed to have spread through prehistoric human migrations from the Old World.
By tracing mutations from global genomic data collected thus far— as brilliantly visualized in Nextstrain (https://nextstrain.org/ncov/global) 27—we can estimate that the time to the most recent common ancestor (tMRCA) of SARS-CoV-2 is 1 December 2019, with geographical origin in Wuhan, Hubei province, China.28 While only 76% of the SARS-CoV-2 genome is similar to SARS-CoV-1, a staggering 96% of it is identical to a bat CoV genome, indicating bat CoV as the closest inter-species ancestor.29 The difference is at the spike protein, which more closely resembles the spike of a pangolin CoV.30 Thus, we can deduce that SARS-CoV-2 jumped from bats as the natural host (CoVs are not deadly to bats), infecting pangolins as a probable intermediate host (which kills them), before mutating further to infect humans.31,32 The virus that was sampled in Wuhan on 5 January, 2020 (isolate name Wuhan/WH04/2020) is the closest to the bat ancestor — this lineage was designated as clade B. Yet, the earliest dated SARS-CoV-2 sample on 26 December, 2019 (Wuhan-Hu-1/2019, now used as the genomic reference) has two distinct mutations (position 8,782 and 28,144), pushing the lineage further away from the ancestor — this type forms the root of SARS-CoV-2 phylogeny in humans and is designated as clade A. From this early bifurcation in Wuhan, both clades A and B were exported outside of Hubei province before spreading to Asia and all over the world.
Several efforts have been made to classify SARS-CoV-2 further down the line. One paper postulated divergence into three types,33 albeit with a debatable method,34 and one preprint even defined sixteen types.35 GISAID at one time established four clades: ‘S,’ ‘G,’ ‘V,’ and ‘Others,’ as shown in the cladogram in Fig. 4.36 Rambaut et al. proposed a dynamic system (the Pangolin lineage, https://pangolin.docs.cog-uk.io) to assign new numbers for every novel mutations, which is robust but resulted in short-lived labels.37 A growing number of ‘types’ are understandable due to the rapidly expanding sampling size, but over-adding taxonomy names is impractical as time passes, not to mention differing proposed names. Unfortunately, there is no universal way to classify viruses below the species level, e.g. HIV-1 ‘subtypes’ have no actual hierarchical relation with ‘subtypes’ in influenza viruses. This prompted a consensus to use a ‘year-clade’ nomenclature for SARS-CoV-2 38: clade A and B were designated 19A and 19B, respectively. Clade 19B— the ancestor —spread first in China on December 2019 albeit in minor prevalence, while 19A— the root clade —spread majorly in China and throughout the rest of Asia. A mutation in spike (D614G) occurred in clade 19A during the massive outbreak in Europe on February 2020, creating clade 20A that quickly spread to all continents and predominates in the current pandemic. From here, a new clade is assigned if it reaches 20% global prevalence. Clade 20A thus branched on late February into clade 20B in Europe (Belgium, Sweden) and 20C into North America.37,39 Thus, a total of five clades can be identified so far, and this nomenclature is now used in Nextstrain (see Fig. 5) and will be used hereafter.
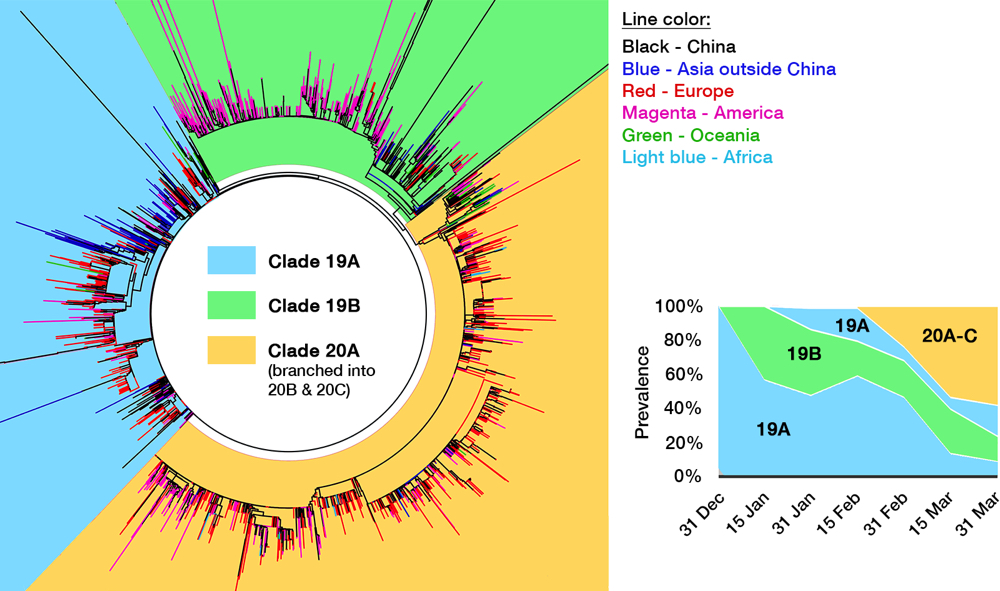
Fig 4. The genomic epidemiology of SARS-CoV-2. While the figure is based on a GISAID report from 12 May, 2020, clade labeling was updated based on the Nextstrain new ‘year-clade’ classification.36 See the main text for clade definition and geographical details. Inset: Clade 20A (and its branch, 20B and 20C) emerged on late February and have gradually supplanted the earlier clades.
When one viral isolate from a place with a mixed population of viruses (e.g. China) is by chance introduced into a new and susceptible place (e.g. Europe), that particular isolate will expand predominantly, creating a viral population dissimilar to the source. This is called the founder effect, which is most likely the driving force behind the varying clade distribution of SARS-CoV-2 between countries. Nevertheless, COVID-19 prevalence and mortality rate broadly differ geographically. For example, CFR is evidently three times higher outside of China (15.2%) compared to within (5.6%).40 It is thus tempting to deduce that due to selection pressure, mutations flourished in SARS-CoV-2 after being exported out from Hubei province, producing more virulent or pathogenic mutants. As discussed above, hotspot mutations are indeed linked to a higher virulence in laboratory tests, especially the D614G spike mutation that makes up clade 20A, whose global prevalence is steadily increasing (see inset of Fig. 4).41 However, the number of infections and CFR are affected by a myriad of factors, including different age demographics between countries,42 polarizing degrees of mitigation policies, disparities in health care facilities,43 and even the inherently varied genetics of human population which may affect initial herd immunity.41,44 Not to mention the complex socioeconomical facet — CFR is tipped to a particular group due to racial and economic inequities.45,46 Until there is a definitive proof that any specific mutation of SARS-CoV-2 makes it to be more virulent in actual human infection, we can only attribute the significant rise of clade 20A to the founder effect47 — the same hypothesis was indeed proven for other viral outbreaks, e.g. HIV-1 and the Ebola virus.
SARS-CoV-2 dissemination in Southeast Asia
It is therefore of interest to investigate the entry and early dissemination of COVID-19 in SEA, to characterize the founder clade and resulting SARS-CoV-2 lineages in the region. The first case in SEA was reported in Thailand on 13 January, from a Wuhan resident visiting Bangkok.48 The first domestic case in Thailand was reported on 31 January from a Thai taxi driver, with source traced to a Chinese passenger.49 Similarly, the first case in Singapore was dated 23 January from a Wuhan tourist, and the first community cluster was reported on 4 February at a local shop catering to a Chinese tour group.50 The outbreak in Singapore then spread into Malaysia — the first report was on 25 January from three China travelers arriving via Singapore, and the first with a Malaysian on 4 February, through a recent trip to Singapore.51 A large outbreak then occurred on 27 February due to an Islamic gathering event known as Tablighi Jamaat in Kuala Lumpur,52 which lead to the first case in Brunei Darussalam on 9 March from a Brunei citizen who attended the event.53
Vietnam reported its first imported case, a father-and-son from Wuhan on 22 January — the case report was then promptly published as the first scientific evidence of human-to-human transmission of COVID-19 outside China.54 The first domestic case was announced on 1 February, with contact traced from the first two cases,55 and the next cluster of infection mostly traced back to Wuhan. Despite directly bordering China, domestic outbreak in Vietnam has been successfully avoided and the incidence rate has been tightly suppressed. Vietnam aptly received global praise for its effective mitigation response even with a low-cost model — similar to their success in handling the 2003 SARS outbreak.56 Vietnamese researchers also managed to sequence viral RNA from a great number of clinical samples, as exhibited in GISAID genome library.
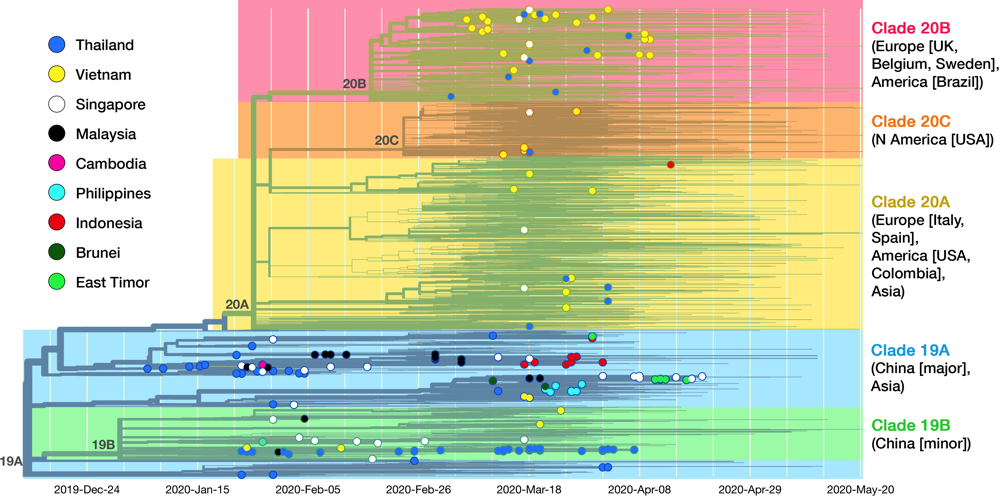
Fig 5. This is the complete cladogram of SARS-CoV-2 with isolates from SEA nations shown in colored nodes. Genomic isolates from all over the world (4305 samples in total) were analyzed and placed in an evolutionary tree. Date is shown on the x axis. The tree is rooted to clade 19A (reference isolate Wuhan-Hu-1/2019). Starting from the two clades (19A, 19B) birthed in Hubei province, the virus diverged into three clades (20A, 20B, 20C) in Europe and America. See text for details on clade dissemination in SEA. Data taken from Nextstrain, sample date ranged until 20 May 2020.13
As of May 2020, the GISAID repository has received 210 SARS-CoV-2 whole-genome sequences from SEA region, with Thailand, Vietnam, and Singapore contributed the most. Fig. 5 shows the latest worldwide cladogram with samples from SEA in colored nodes. Fig. 6 shows the approximate location of data sources from each SEA country and their respective clade distribution. Isolates from Thailand and Singapore are mostly split rather equally into clade 19A and 19B, which represents the Wuhan route of entry. Early Vietnamese samples also came from those two Chinese clades, but recent sequencing efforts in Vietnam, as well as from Thailand and Singapore, identified numerous clade 20B isolates. This indicates a rather concerning re-entry of cases from Europe back into SEA. Meanwhile, almost all Malaysian isolates so far were shown to be clade 19A, as well as Brunei.
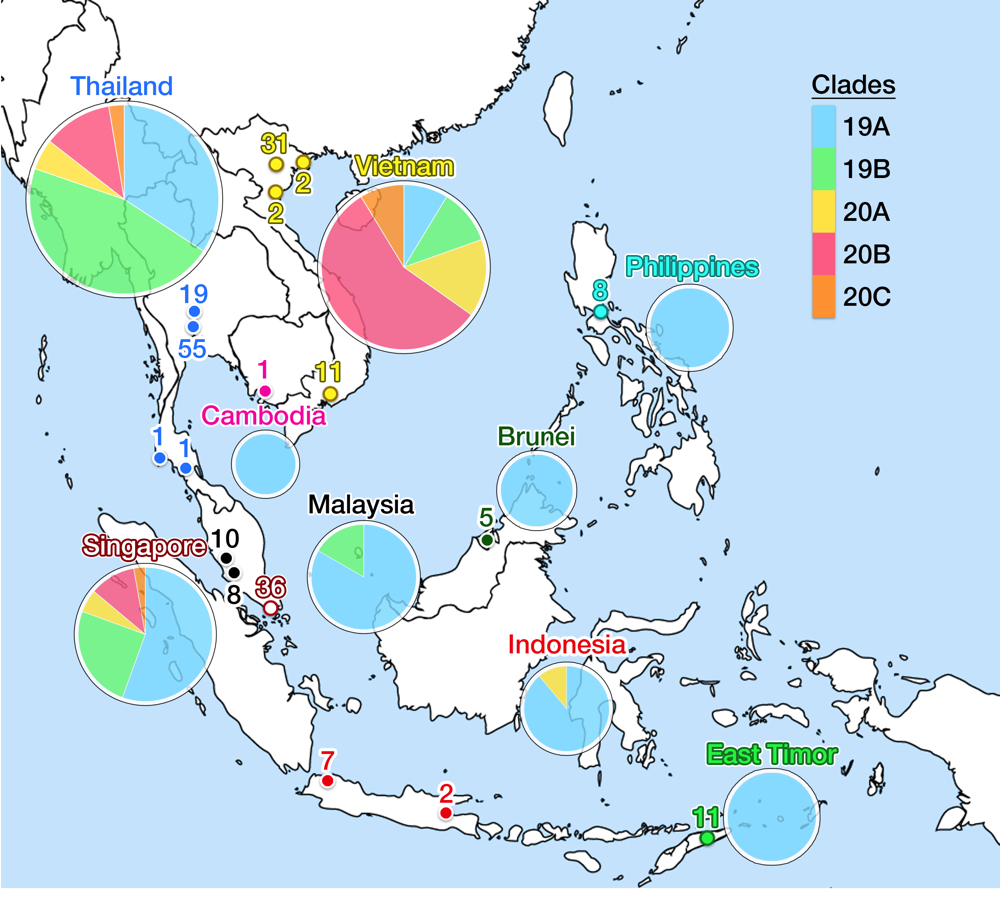
Fig 6. This figure shows the geographical location of SARS-CoV-2 genomic isolates in SEA and clade distribution for each country. Places identified as isolate sources are mapped in colored nodes (see Fig. 5 for node color legend). The numbers above the node signify the amount of samples from respective locations. Clade variance is shown in a pie chart below the name of the country; the chart diameter comparatively indicates the amount of isolate sampled. Clade 19A is the major clade in most countries, but more recent isolates from Thailand, Vietnam and Singapore show a more varied population with possible re-entry of cases from Europe. No data is available for Myanmar and Laos. Data is taken from the GISAID library and Nextstrain, updated on 4 June 2020.13
Perennially busy transit between China and SEA has established the main route of SARS-CoV-2 entry into Thailand and Singapore, but not necessarily for subsequent countries. The first imported case into Cambodia was reported on 27 January from a Wuhan resident,57 but the first domestic case was announced on 7 March in Siem Reap, traced back to a Japanese national.58 The first case in the Philippines also came from Wuhan on 30 January, but a domestic case was not reported until 6 March from a Filipino returning from Japan. Community outbreak in Metro Manila was confirmed on the same day.59 On 2 March, two Indonesians were confirmed to be positive in Jakarta, whose contact was traced back to a Japanese national. Imported cases were not discovered first in Indonesia, and the first domestic reports lagged when compared to other SEA countries. Regardless, COVID-19 prevalence in Indonesia rapidly shot up to 28,818 cases as of 4 June to become the second highest in SEA after Singapore, with a menacing CFR of 6.1%, far above all other SEA countries and ranked fifth in Asia.60 Numbers may be severely underestimated as Indonesia has one of the worst testing rates in the world (1,225 tests per million people).61 There is a recent shift in the outbreak epicenter from the capital to East Java— presumably due to the flow of people returning to their hometowns at the end of Ramadan —causing further concern.
All isolates from Cambodia, Philippines, and Indonesia (except one) were grouped into clade 19A (the original Wuhan clade), which also agrees with the genomic epidemiology of one of the source countries, Japan.62 The most recent isolate from Indonesia was identified as clade 20A, which again indicated possible re-entry of European cases. Thus, more recent data is critically needed, but the amount of isolate sequencing from these three countries, despite their size and population, are awfully lagging behind the three SEA front-runners. The Indonesian outbreak then spread to cause the first community case in East Timor on 21 March.63 Admirably, East Timor has already uploaded eleven isolates into GISAID: all were clade 19A. The two last SEA countries to report COVID-19 case were Myanmar and Laos. The first Laotian caught the virus from the outbreak in Thailand, reported on 24 March.64 On the same day, Myanmar reported two of its citizen testing positive, each returning from United States and Britain which signify another re-entry event.65 Sequencing reports from these two countries were unavailable at the time of writing of this article.
Concluding remarks
A successful vaccine development hinges on four traits of the virus: virulence, viral fitness, diversity, and evolutionary dynamic. Despite its high virulence and fitness, SARS-CoV-2 has exhibited limited diversity and slow evolution based on genomic evidence so far. But the pandemic is still ongoing; data collection needs to be continuously sustained. We have to be critical of sampling biases as well — proportionally more data is produced in the northern hemisphere.37 Data from SEA nations and other developing countries are similarly indispensable. Even though vaccine R&D is essentially an arms race against viral evolution, it cannot be rushed; premature deployment of ineffectual or unsafe vaccine will only erode public trust. Misinformation and false skepticism have put a lasting damage to vaccination campaign before, as seen for polio and measles virus.66 We cannot afford to have recurring waves of COVID-19, and two thirds of the populace having to acquire immunity naturally to stop the pandemic.67 Vaccines have halted pandemics of the past. Could one vaccine potentially be used in all parts of the world to stop COVID-19? The answer should be a resounding yes, and we have to strive to achieve it.
The author would like to thank Prof. Yoshitake Hayashi, Dr. Morgane Rolland, Dr. Tomohiro Kotaki, and Dr. Yan Mardian for the fruitful discussions and help toward writing this article.
5 June, 2020
References
- 1 World Health Organization (WHO). 2 June, 2020. Draft landscape of COVID-19 candidate vaccines. https://www.who.int/who-documents-detail/draft-landscape-of-covid-19-candidate-vaccines. (Accessed 4 June, 2020).
- 2 World Health Organization (WHO). 20 January, 2020. Novel coronavirus (2019-nCoV) situation report-1. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200121-sitrep-1-2019-ncov.pdf?sfvrsn=20a99c10_4. (Accessed 25 May, 2020).
- 3 Anonymous. 2020. A COVID-19 vaccine might be ready within 18 months. But what happens then? Bill & Melinda Gates Foundation. https://www.gatesfoundation.org/TheOptimist/Articles/coronavirus-vaccine-development-gavi. (Accessed 26 May, 2020).
- 4 Burton DR, Walker LM. 22, April, 2020. Rational vaccine design in the time of COVID-19. Cell Host Microbe. 2020;27(5):695-698. doi:10.1016/j.chom.
- 5 Lan J, Ge J, Yu J, et al. 2020. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 581(7807):215-220. doi:10.1038/s41586-020-2180-5
- 6 Smith TRF, Patel A, Ramos S, et al. 2020. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat Commun. 11(1):2601. doi:10.1038/s41467-020-16505-0
- 7 Xie L, Sun C, Luo C, et al. February 2020. SARS-CoV-2 and SARS-CoV spike-RBD structure and receptor binding comparison and potential implications on neutralizing antibody and vaccine development. bioRxiv. 2020.02.16.951723. doi:10.1101/2020.02.16.951723
- 8 Bwhana PG. Jokowi: we need to develop our own COVID-19 vaccine. May 20, 2020. Tempo. https://en.tempo.co/read/1344441/jokowi-we-need-to-develop-our-own-covid-19-vaccine. (Accessed 4 June, 2020).
- 9 Kittisilpa J, Wongcha-um P. May 25, 2020. Thai researcher eyes affordable, accessible coronavirus vaccine for SE Asia. Reuters. https://www.reuters.com/article/us-health-coronavirus-thailand-vaccine/thai-researcher-eyes-affordable-accessible-coronavirus-vaccine-for-se-asia-idUSKBN2311DE. (Accessed 26 May, 2020).
- 10 Duffy S. 2018. Why are RNA virus mutation rates so damn high? PLoS Biol. 16(8):e3000003. doi:10.1371/journal.pbio.3000003
- 11 Hanley KA. 2011. The double-edged sword: how evolution can make or break a live-attenuated virus vaccine. Evol Educ Outreach. 4(4):635-643. doi:10.1007/s12052-011-0365-y
- 12 Fitzsimmons WJ, Woods RJ, McCrone JT, et al. 2018. A speed–fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 16(6):e2006459. doi:10.1371/journal.pbio.2006459
- 13 Nextstrain. Genomic epidemiology of novel coronavirus: global subsampling. Nextstrain. https://nextstrain.org/ncov/global. 30 May, 2020. (Accessed 4 June, 2020).
- 14 Dearlove B, Lewitus E, Bai H, et al. January, 2020. A SARS-CoV-2 vaccine candidate would likely match all currently circulating strains. bioRxiv. 2020.04.27.064774. doi:10.1101/2020.04.27.064774
- 15 Yuan M, Wu NC, Zhu X, et al. 2020. A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV. Science. 368(6491):eabb7269. doi:10.1126/science.abb7269
- 16 Global Initiative on Sharing All Influenza Data (GISAID). 25 May, 2020. Genomic epidemiology of novel coronavirus: global subsampling. https://www.gisaid.org/epiflu-applications/next-hcov-19-app/. (Accessed 4 June, 2020).
- 17 Global Initiative on Sharing All Influenza Data (GISAID). 13 April, 2020. Real-time tracking of influenza A/H3N2 evolution using data from GISAID. https://nextstrain.org/flu/seasonal/h3n2/ha/2y?l=clock. (Accessed 31 May, 2020).
- 18 Surdel MC. 2014. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog. 10(7):e1003565. doi:10.1371/journal.ppat.1004342
- 19 Peck KM, Lauring AS. 2018. Complexities of viral mutation rates. Sullivan CS, ed. J Virol. 92(14):e01031-17. doi:10.1128/jvi.01031-17
- 20 Lau EHY, Hsiung CA, Cowling BJ, et al. 2010. A comparative epidemiologic analysis of SARS in Hong Kong, Beijing and Taiwan. BMC Infect Dis. 10:50. doi:10.1186/1471-2334-10-50
- 21 Cotten M, Watson SJ, Zumla AI, et al. 2014. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. MBio. 5(1):e01062-13. doi:10.1128/mBio.01062-13
- 22 Vijgen L, Keyaerts E, Moës E, et al. 2005. Complete Genomic Sequence of Human Coronavirus OC43: Molecular Clock Analysis Suggests a Relatively Recent Zoonotic Coronavirus Transmission Event. J Virol. 79(3):1595-1604. doi:10.1128/jvi.79.3.1595-1604.2005
- 23 Ou J, Zhou Z, Dai R, et al. March, 2020. Emergence of RBD mutations from circulating SARS-CoV-2 strains with enhanced structural stability and higher human ACE2 receptor affinity of the spike protein. bioRxiv. 2020.03.15.991844. doi:10.1101/2020.03.15.991844
- 24 Pachetti M, Marini B, Benedetti F, et al. 2020. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. 18(1):179. doi:10.1186/s12967-020-02344-6
- 25 Korber B, Fischer W, Gnanakaran SG, et al. January, 2020. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv. 2020.04.29.069054. doi:10.1101/2020.04.29.069054
- 26 Sagulenko P, Puller V, Neher RA. 2018. TreeTime: maximum-likelihood phylodynamic analysis. Virus Evol. 4(1):vex042-vex042. doi:10.1093/ve/vex042
- 27 Hadfield J, Megill C, Bell SM, et al. 2018. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 34(23):4121-4123. doi:10.1093/bioinformatics/bty407
- 28 Lu J, Plessis L du, Liu Z, et al. April, 2020. Genomic epidemiology of SARS-CoV-2 in Guangdong province, China. Cell. S0092-8674(20)30486-4. doi:10.1016/j.cell.2020.04.023
- 29 Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. 2020. The proximal origin of SARS-CoV-2. Nat Med. 26(4):450-452. doi:10.1038/s41591-020-0820-9
- 30 Lam TT-Y, Shum MH-H, Zhu H-C, et al. 2020. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature. doi:10.1038/s41586-020-2169-0
- 31 Zhang YZ, Holmes EC. 2020. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell. 181(2):223-227. doi:10.1016/j.cell.2020.03.035
- 32 Gao W-H, Lin X-D, Chen Y-M, et al. 2020. Newly identified viral genomes in pangolins with fatal disease. Virus Evol. 6(1):veaa020. doi:10.1093/ve/veaa020
- 33 Forster P, Forster L, Renfrew C, Forster M. 2020. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci. 117(17):9241 LP – 9243. doi:10.1073/pnas.2004999117
- 34 Mavian C, Pond SK, Marini S, et al. May, 2020. Sampling bias and incorrect rooting make phylogenetic network tracing of SARS-COV-2 infections unreliable. Proc Natl Acad Sci. 202007295. doi:10.1073/pnas.2007295117
- 35 Júnior IJM, Polveiro RC, Souza GM, Bortolin DI, Sassaki FT, Lima ATM. April, 2020. The global population of SARS-CoV-2 is composed of six major subtypes. bioRxiv. 2020.04.14.040782. doi:10.1101/2020.04.14.040782
- 36 Global Initiative on Sharing All Influenza Data (GISAID). 4 April, 2020. Full genome tree of all outbreak sequences. https://www.gisaid.org/hcov-19-analysis-update. (Accessed 29 May, 2020).
- 37 Rambaut A, Holmes EC, Hill V, et al. April, 2020. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv. 2020.04.17.046086. doi:10.1101/2020.04.17.046086
- 38 Hodcroft EB, Hadfield J, Neher RA, Bedford T. 3 June, 2020. Year-letter genetic clade naming for SARS-CoV-2 on nextstrain.org. Nextstrain. https://virological.org/t/year-letter-genetic-clade-naming-for-sars-cov-2-on-nextstain-org/498. (Accessed 4 June, 2020).
- 39 Eden JS, Rockett R, Carter I, et al. 2020. An emergent clade of SARS-CoV-2 linked to returned travellers from Iran. Virus Evol. 6(1):veaa027. doi:10.1093/ve/veaa027
- 40 Baud D, Qi X, Nielsen-Saines K, Musso D, Pomar L, Favre G. June, 2020. Real estimates of mortality following COVID-19 infection. Lancet Infect Dis. doi:10.1016/S1473-3099(20)30195-X
- 41 Bhattacharyya C, Das C, Ghosh A, et al. May, 2020. Global spread of SARS-CoV-2 subtype with spike protein mutation D614G is shaped by human genomic variations that regulate expression of TMPRSS2 and MX1 genes. bioRxiv. 2020.05.04.075911. doi:10.1101/2020.05.04.075911
- 42 Li H, Wang S, Zhong F, et al. February, 2020. Age-dependent risks of incidence and mortality of COVID-19 in Hubei province and other parts of China. medRxiv. 2020.02.25.20027672. doi:10.1101/2020.02.25.20027672
- 43 He Y. Illness and fatality risks of COVID-19 of general public in Hubei provinces and other parts of China. February, 2020. medRxiv. 2020.02.25.20027672. doi:10.1101/2020.02.25.20027672
- 44 Grifoni A, Weiskopf D, Ramirez SI, et al. May, 2020. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. doi:10.1016/j.cell.2020.05.015
- 45 Abedi V, Olulana O, Avula V, et al. 2020. Racial, economic and health inequality and COVID-19 infection in the United States. medRxiv. 2020.04.26.20079756. doi:10.1101/2020.04.26.20079756
- 46 Yancy CW. COVID-19 and African Americans. 15 April, 2020. JAMA. 323(19):1891-1892. doi:10.1001/jama.2020.6548
- 47 Farkas C, Fuentes-Villalobos F, Garrido JL, Haigh JJ, Barría MI. April, 2020. Insights on early mutational events in SARS-CoV-2 virus reveal founder effects across geographical regions. bioRxiv. 2020.04.09.034462. doi:10.1101/2020.04.09.034462
- 48 Cheung E. Wuhan pneumonia: Thailand confirms first case of virus outside China. 13 January, 2020. South China Morning Post. https://www.scmp.com/news/hong-kong/health-environment/article/3045902/wuhan-pneumonia-thailand-confirms-first-case. (Accessed 4 June, 2020).
- 49 Anonymous. 31 January, 2020. สธ.แถลง พบคนขับแท็กซี่ ติดไวรัสโคโรน่า เป็นคนไทยรายแรก ไม่มีประวัติไปจีน (MOPH announces taxi driver infected with coronavirus—first Thai case with no records of travelling to China). Thai Rath. https://www.thairath.co.th/news/local/bangkok/1761053. (Accessed 3 June, 2020).
- 50 Lien C-A, Khalik S. 4 February, 2020. Coronavirus: S’pore reports first cases of local transmission; 4 out of 6 new cases did not travel to China. The Straits Times. https://www.straitstimes.com/singapore/coronavirus-spore-reports-first-cases-of-local-transmission-4-out-of-6-new-cases-did-not. (Accessed 3 June, 2020).
- 51 Anonymous. 4 February, 2020. First case of Malaysian positive for coronavirus. Bernama. https://www.bernama.com/en/general/news_coronavirus.php?id=1811373. (Accessed 3 June, 2020).
- 52 Ananthalakshmi A, Sipalan J. 18 March, 2020. How mass pilgrimage at Malaysian mosque became coronavirus hotspot. Reuters. https://www.reuters.com/article/us-health-coronavirus-malaysia-mosque/how-mass-pilgrimage-at-malaysian-mosque-became-coronavirus-hotspot-idUSKBN2142S4. (Accessed 3 June, 2020).
- 53 Ministry of Health Brunei Darussalam. 9 March, 2020. Detection of the first case of COVID-19 infection in Brunei Darussalam. http://www.moh.gov.bn/Lists/Latest news/NewDispForm.aspx?ID=366. (Accessed 4 June, 2020).
- 54 Phan LT, Nguyen T V., Luong QC, et al. Importation and human-to-human transmission of a novel coronavirus in Vietnam. N Engl J Med. 2020;382(9):872-874. doi:10.1056/NEJMc2001272
- 55 Anonymous. 4 February 4, 2020.Bệnh nhân viêm phổi thứ ba xuất viện (Third pneumonia patient discharged). VnExpress. https://vnexpress.net/benh-nhan-viem-phoi-thu-ba-xuat-vien-4050123.html. (Accessed 4 June, 2020).
- 56 Reed J. 24, March, 2020. Vietnam’s coronavirus offensive wins praise for low-cost model. Financial Times. https://www.ft.com/content/0cc3c956-6cb2-11ea-89df-41bea055720b. (Accessed 4 June, 2020).
- 57 Sithirith M. 2020. COVID-19 in Cambodia: a double-edged sword and its triple effects on Democracy. CSEAS Newsl. 2020;(78).
- 58 Tostevin M, Williams A. 7 March, 2020.First Cambodian tests positive for coronavirus. Reuters. https://www.reuters.com/article/us-health-coronavirus-cambodia/first-cambodian-tests-positive-for-coronavirus-idUSKBN20U0HW. (Accessed 4 June, 2020).
- 59 Punzalan J. 6 March, 2020.Philippines’ new coronavirus cases now at 5, including potential local transmission. ABS-CBN News. https://news.abs-cbn.com/news/03/06/20/new-coronavirus-case-in-philippines-possible-local-transmission. (Accessed 4 June, 2020).
- 60 Gugus Tugas Percepatan Penanganan COVID-19. 2020. Peta Sebaran Kasus per Provinsi. Jakarta: Gugus Tugas Percepatan Penanganan COVID-19. https://covid19.go.id/peta-sebaran. (Accessed 4 June, 2020).
- 61 Anonymous. 2 June, 2020. Indonesia’s daily coronavirus testing consistently on target. Jakarta Globe. https://jakartaglobe.id/news/indonesias-daily-coronavirus-testing-consistently-on-target. (Accessed 4 June, 2020).
- 62 National Institute of Infectious Diseases. 2020. An Epidemiological Study of the SARS-CoV-2 Genome in Japan. Tokyo: NIID. https://www.niid.go.jp/niid/en/basic-science/467-genome/9598-genome-2020-1e.html. (Accessed 4 June, 2020).
- 63 Cruz N Da, Ungku F. 21 March, 2020. East Timor confirms first case of coronavirus: health ministry. Reuters. https://www.reuters.com/article/us-health-coronavirus-timor/east-timor-confirms-first-case-of-coronavirus-health-ministry-idUSKBN2180BI. (Accessed 4 June, 2020).
- 64 Wongcha-um P, Elgood G. 24 March, 2020. Laos records first two coronavirus cases: Thai media. Reuters. https://www.reuters.com/article/us-health-coronavirus-laos/laos-records-first-two-coronavirus-cases-thai-media-idUSKBN21B0X2. (Accessed 4 June, 2020).
- 65 Fullick N. 24 March, 2020. Myanmar reports first cases of coronavirus. Reuters. https://www.reuters.com/article/us-health-coronavirus-myanmar/myanmar-reports-first-cases-of-coronavirus-idUSKBN21B0HB. (Accessed 4 June, 2020).
- 66 Trogen B, Oshinsky D, Caplan A. May, 2020. Adverse consequences of rushing a SARS-CoV-2 vaccine: implications for public trust. JAMA. doi:10.1001/jama.2020.8917
- 67 Randolph HE, Barreiro LB. 2020. Herd immunity: understanding COVID-19. Immunity. 52(5):737-741. doi:10.1016/j.immuni.2020.04.012
Youdiil Ophinni obtained his M.D from University of Indonesia in 2011 and earned his Ph.D in Virology from Kobe University in 2018. He is currently a postdoctoral researcher at the Ragon Institute of MGH, MIT and Harvard.
Citation
Youdiil Ophinni. 2020. “SARS-CoV-2 Mutation and Dissemination in Southeast Asia: Implications for a Prospective Vaccine” CSEAS NEWSLETTER, 78: TBC.




